About MLD
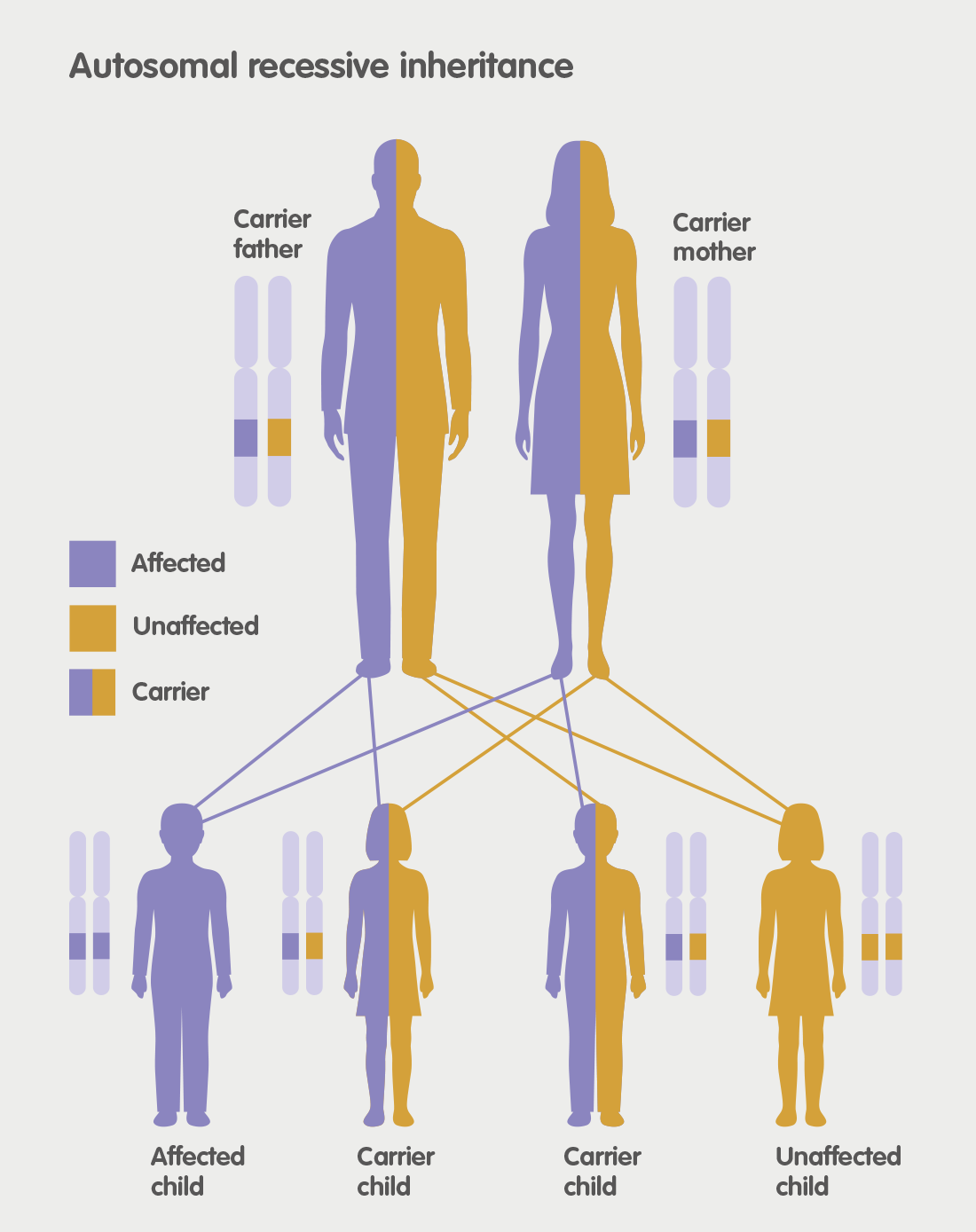
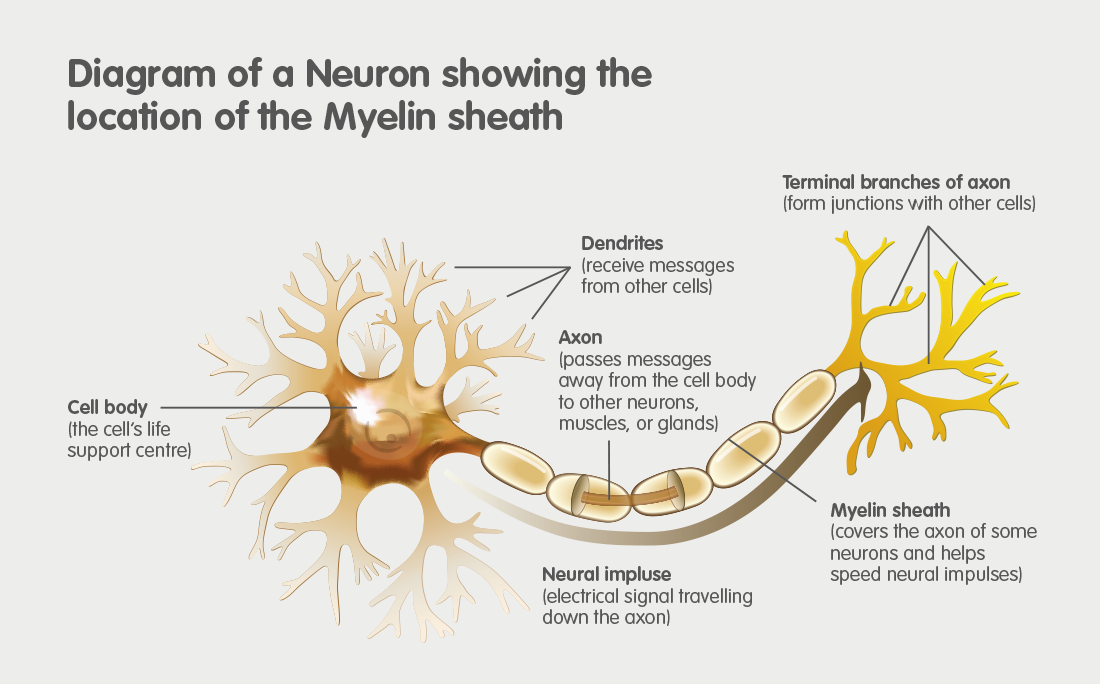
MLD is an acronym for Metachromatic Leukodystrophy. MLD is an autosomal recessive genetic disorder which at the moment has no cure. MLD is directly caused by a deficiency of the enzyme Arylsulfatase-A. Without this enzyme sulfatides build up, eventually destroying the myelin sheath of the nervous system. The myelin sheath is a fatty covering that protects nerve fibres. Without it, the nerves in the brain and the peripheral nerves cease to function properly.
What is the incidence of MLD
There are many forms of Leukodystrophy, but Metachromatic Leukodystrophy (MLD) is one of the most common forms. The incidence of MLD is now estimated to occur in 1 case in 40,000 live births. However, with modern diagnostic tools such as MRI Scans and genetic sequencing, it means that there are fewer incorrect diagnoses and it is possible that the incidence may prove to be higher.
Types of MLD
Like many other genetic disorders that affect lipid metabolism, there are several forms of MLD, which are generally classified as Late-Infantile, Juvenile (sometimes broken into Early-Juvenile and Juvenile) and, finally, Adult-Onset. In the late-infantile form, which is the most common form of MLD (50-60%), affected children begin having difficulty walking after the first year of life, usually at 15–24 months. Juvenile MLD has an onset between 3 and 10 years of age, usually beginning with impaired school performance. Adult-Onset MLD is the rarest form and commonly begins after age 16 and, in the initial stages, is often mis-diagnosed as a psychiatric disorder because of personality changes.
Late Infantile MLD
The most common initial signs and symptoms in children who develop MLD during the first 2 years of life are, after a period of normal growth and development, abnormal or erratic movements and other changes in the way they move particularly when they are crawling or walking. Unfortunately, as MLD is such a rare disease and health professionals generally see less than one or two cases in their career, these children are frequently diagnosed as cerebral palsy as they initially have problems with walking and toe walking. However, with MLD there is also deterioration in other developmental skills e.g. loss of speech. Once these symptoms become noticeable they can progress rapidly over a few months but with periods of apparent stabilisation.
Although children with Late-infantile MLD do generally learn to walk, the movement in the limbs becomes abnormal and, as the muscles become rigid, walking ability diminishes. Speech is also affected and, as many parents have noted, this is one of the unkindest symptoms—not hearing their children’s voices again. As time goes by, limbs can become painful as contractures develop. Fine motor skills are affected, for instance writing, drawing etc. Difficulties in feeding occur and weight loss becomes a significant problem.
Many children are tube fed to overcome this. Ultimately, mobility and speech is totally lost and the child becomes bedridden. This is accompanied by a progressive cognitive decline, restricting interaction and understanding and, eventually, leading to dementia. Sight is also lost and seizures become a significant problem. Unfortunately, once symptoms have appeared it is too late for any treatment. Gene Therapy is now available at specialist centres on the NHS. This treatment has proved effective, but only with children who are pre-symptomatic.
Early-Juvenile and Late-Juvenile MLD
Children who start to develop symptoms from around 3 to 16 years of age often show initial impairments in fine motor skills and concentration, and may develop behavioural problems, particularly at school.
They begin to find it difficult keeping up with their peers and may sometimes be referred to a child psychologist or psychiatrist as a psychiatric condition or depression is suspected. As with Late-infantile the MLD diagnosis is often protracted with many wrong suggestions. Unfortunately, this delay often means that any treatment so far available becomes impossible as the symptoms are too far advanced.
In the early stages children often suffer increasing difficulties with movement and co-ordination and walking. They can develop slurred speech and suffer incontinence. The situation may stabilise for months or even years in some cases, but the reason for this is not yet known. However, as symptoms advance, individuals develop signs of neurological involvement such as involuntary flexion or extension of the arms and legs, tremor, muscle rigidity and, eventually, they lose the ability to walk. This is followed by a decline in motor and cognitive function and, unfortunately, the final stage of the disease follows that of the Late-infantile form of MLD, but generally over a longer period of time.
Currently, Gene Therapy is available at specialist centres on the NHS. This treatment has proved effective, but only for children with Early-Juvenile MLD who are pre-symptomatic or early-symptomatic, that is having very mild symptoms. A clinical trial is currently being carried out in Milan for children who are pre-symptomatic or early-symptomatic with Late-Juvenile MLD.
Adult-Onset MLD
Adult MLD is the rarest form of Metachromatic Leukodystrophy and, generally, the most difficult to diagnose. Early signs and symptoms in adults who develop MLD are frequently a sudden and unexpected poor performance at school or university, or in other academic work. Initially, this decline can be very slow and almost imperceptible. Along with the slow decline in intellectual and cognitive abilities the sufferers personalities change and they can become emotionally unstable and experience lapses in memory.
Again, unfortunately, this is normally initially misdiagnosed as a psychiatric illness, frequently schizophrenia and medication is given which, generally, exacerbates the problems. Getting a person suffering from Adult-MLD away from a psychiatrist and to a neurologist seems to be the only hope of getting the correct diagnosis. Many clinicians believe that Adult-Onset MLD is currently much under-diagnosed.
Sufferers of Adult-Onset MLD can benefit most from Bone Marrow or Stem Cell Transplant as their degeneration is at a much slower pace. Indeed, both in the UK and the USA there are now a fair number of cases which have been extremely successfully with the disease showing no progression some 30 years later. However, this is dependent on the sufferer being pre-symptomatic or only having mild symptoms e.g. no physical problems at the time of the BMT/SCT. Early diagnosis is the key here.
Without intervention, as the peripheral nervous system becomes involved, movements may become clumsy and they may become incontinent. Paralysis of the arms and legs develops progressively.
.